L’amylose AL
L’amylose AL (A pour amylose et L pour chaînes légères d’immunoglobuline) est une pathologie rare et grave (médiane de survie d’un an sans traitement), se manifestant par des atteintes d’organes très variées et parfois sévères. L’évolution des traitements ces dernières années a permis une amélioration majeure du pronostic, conditionnée par une prise en charge rapide avant la constitution d’atteintes sévères, en particulier cardiaque. Un diagnostic précoce est donc un facteur majeur pour le pronostic des patients atteints et les généralistes sont en première ligne pour le faire.
Par Sammara Chaubard, Arnaud Jaccard, service d’Hématologie clinique et de Thérapie cellulaire, Centre de référence amylose AL et autres maladies par dépôt d’immunoglobulines monoclonales, CHU Limoges.
Épidémiologie
L’amylose AL est une pathologie 5 fois moins fréquente que le myélome, avec environ 600 nouveaux cas par an en France. L’âge moyen au diagnostic est 65 ans, mais les adultes plus jeunes peuvent également être touchés avec environ 10% de patients diagnostiqués avant 50 ans.
Physiopathologie
L’amylose AL est liée à la sécrétion de chaines légères monoclonales d’immunoglobulines qui, du fait de leur structure tertiaire instable, se polymérisent sous forme de fibrilles amyloïdes qui se déposent dans le milieu extracellulaire des différents organes. Ces dépôts sont à l’origine des manifestations cliniques de cette pathologie qui sont très variées en fonction des organes atteints.
La prolifération B monoclonale à l’origine de la production des chaînes légères amyloïdogènes est le plus souvent plasmocytaire correspondant à une MGUS (monoclonal gammopathy of unknown significance) ou à un myélome indolent, l’évolution vers un myélome symptomatique étant rare. Elle est dans moins de 10% des cas lymphocytaire ou lympho-plasmocytaire, l’immunoglobuline monoclonale étant alors plutôt d’isotype IgM.
Il existe d’autres formes d’amylose :
– héréditaires liées à la mutation du gène de certaines protéines les rendant capables de polymériser sous forme de fibrilles
– secondaires (amylose AA) liées à une inflammation chronique augmentant le taux sérique de la protéine SAA
– par dépôts de β2microglobuline liés à l’augmentation de son taux sérique chez les patients dialysés
– par dépôt de transthyrétine non mutée dans le cœur des patients âgés (amylose TTR sénile).
Clinique
Les atteintes sont multiples et variées, tous les organes hormis le système nerveux central pouvant être atteints (Tableau 1). Les atteintes les plus fréquentes sont cardiaquescardiaques et rénales, et les symptômes initiaux souvent peu spécifiques (asthénie et dyspnée d’effort). Mais certains patients ont des manifestations typiques qu’il faut connaître car elles permettent d’évoquer le diagnostic : macroglossie et hématomes des paupières (Figure 1).
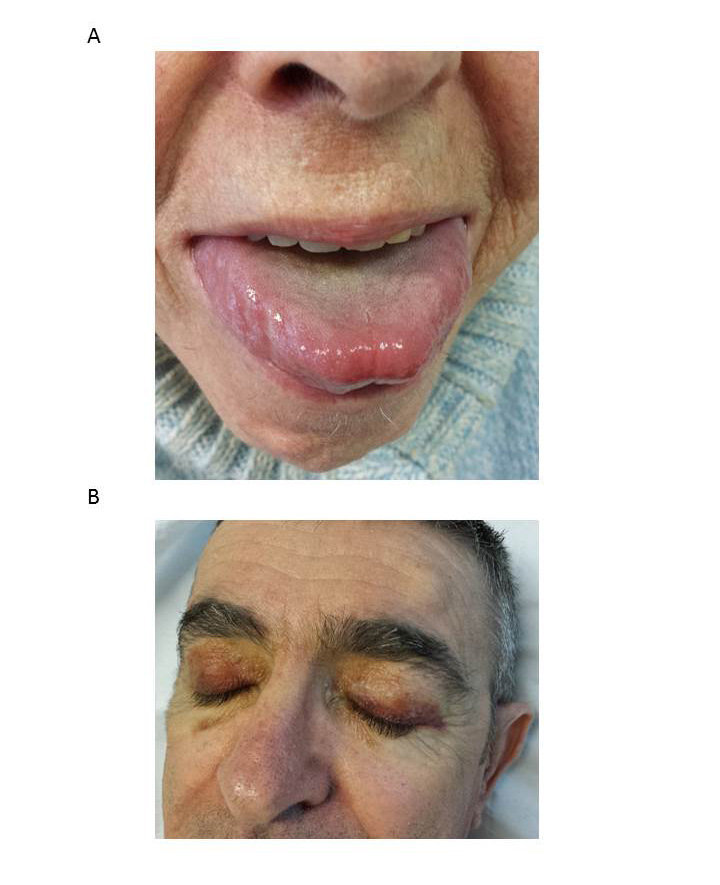
Figure 1 : A - macroglossie ; B - hématomes péri-orbitaires
Il existe également des formes localisées d’amylose AL avec des localisations très variées, paupières, vessie et voies urinaires, larynx et bronches, tube digestif, peau, etc. Ces formes sont le plus souvent liées à des proliférations monoclonales B de bas grade (lymphome de la zone marginale ou plasmocytome extra-osseux), avec dépôt in situ de la protéine monoclonale, à proximité de son lieu de production.
Afin d’éliminer les autres formes d’amylose, on recherche l’existence d’une maladie inflammatoire chronique (Amylose AA) ou d’antécédents familiaux d’amylose héréditaire (par mutation du gène de la transthyrétine ou de la chaine alpha du fibrinogène surtout).
Organe/Tissu | Symptomatologie |
Signes peu spécifiques | Asthénie, amaigrissement, œdèmes, dyspnée |
Neurologique | - Polyneuropathie sensorielle longueur-dépendante, douloureuse: atteinte sensibilité thermo-algique -> paresthésies -> déficit moteur Syndrome du canal carpien - Neuropathie dysautonomique: hypotension orthostatique, perte de la sudation, troubles gastro-intestinaux, surtout diarrhée, dysfonction vésicale, impuissance |
Cardiaque | Cardiomyopathie restrictive d’aggravation progressive: Asthénie -> Dyspnée d’effort |
Rénal | 2/3 cas Atteinte glomérulaire, hypoalbuminémie et œdèmes des membres inférieurs |
Hépatique | Hépatomégalie avec cholestase anictérique Rarement ictère cholestatique: facteur de mauvais pronostic |
Splénique | Splénomégalie avec hyposplénisme |
Digestif | Macroglossie: pathognomonique mais rare (15%) Infiltration muqueuse buccale -> agueusie, xérostomie -> amaigrissement Malabsorption |
Cutané | Purpura péri-oculaire, ecchymoses Papules, nodules, plaques au niveau de la face et partie supérieure tronc Rarement formes bulleuses |
Pulmonaire | - Nodulaire: forme souvent peu évolutive - Interstitielle dans la majorité des cas |
Rhumatologique | Pseudo-hypertrophie musculaire Poly-arthropathies bilatérales et symétriques des doigts (doigt à ressaut), poignets, épaules et genoux Signe de l’épaulette par infiltration des gaines tendineuses des épaules |
Tableau 1. Signes cliniques de l’amylose AL.
Examens complémentaires
Le diagnostic d’amylose AL comporte plusieurs étapes (Tableau 2 et tableau 3) :
– Diagnostic d’amylose par biopsies le plus souvent non invasives (glandes salivaires accessoires et graisse sous-cutanée) et coloration au Rouge Congo qui fait le diagnostic. L’anatomopathologiste doit être entraîné au diagnostic des amyloses et prévenu de la suspicion afin qu’il recherche des dépôts par la coloration du Rouge Congo.
– Typage de l’amylose à l’aide des anticorps spécifiques des différentes formes
– Recherche et caractérisation d’une protéine monoclonale sanguine et/ou urinaire et caractérisation de l’hémopathie responsable : MGUS (monoclonal gammopathy of unknown significance) ou myélome multiple, en général de faible masse tumorale, et quand l’immunoglobuline est une IgM : lymphome de bas grade ou maladie de Waldenström.
Examens biologiques | Résultats |
Electrophorèse et immunofixation des protides sanguins et urinaires Protéinurie des 24h Bandelette urinaire | Pic monoclonal et typage de la protéine monoclonale Protéinurie glomérulaire avec >30% d’albuminurie si atteinte rénale Protéinurie de Bence-Jones Rare hématurie microscopique |
Dosage des chaines légères libres circulantes | Augmentation d’un isotype avec rapport ĸ/λ anormal |
Créatinine+clairance, urée plasmatique, protides, albuminémie | Syndrome néphrotique: protéinurie>3g/24h, protidémie<60g/L et albuminémie<30g/L |
Troponine us, NT-proBNP | Elevées si atteinte cardiaque (importantes pour le dépistage ++++) |
Ferritinémie | Abaissée si saignement digestif |
ASAT, ALAT, GGT, PAL, bilirubine | Cholestase anictérique avec augmentation isolée PAL Rare augmentation bilirubine (mauvais pronostic) |
NFS, bilan de coagulation avec dosage du facteur X | Thrombocytose si asplénie Déficit en facteur X |
Tableau 2. Examens biologiques utiles dans les amyloses AL.
Bilan cardiologique: - ECG: -> microvoltage dans les dérivations périphériques (QRS<0,5cm), -> ondes Q de pseudo-nécrose en V1-V2, -> troubles du rythme auriculaire ou ventriculaire, -> troubles de la conduction - Echographie cardiaque: Cardiopathie restrictive avec hypertrophie des parois cardiaques -> FEVG (altération tardive), -> aspect granité et brillant du myocarde, -> épaisseur du septum inter-ventriculaire en diastole (>12mm) -> doppler: dysfonction diastolique |
Autres examens complémentaires: - Radiographie pulmonaire: épanchement pleural bilatéral, pneumopathie interstitielle - Echographie abdominale: recherche d’une hépato-splénomégalie, reins - Recherche hémopathie associée: -> Myélogramme: pourcentage de plasmocytes médullaires - IRM cardiaque : diagnostic précoce -> cardiopathie hypertrophique -> rehaussement tardif caractéristique en T1 injecté (dépôts) - Holter-ECG: Indispensable si atteinte cardiaque ++++ - Electromyogramme: Atteinte des petites fibres nerveuses |
Tableau 3. Examens complémentaires utiles dans les amyloses AL.
Clinique, evolution et facteurs pronostiques
Les atteintes cliniques sont souvent multiples :
– Atteinte cardiaque : insuffisance cardiaque restrictive responsable d’une dyspnée d’effort s’aggravant progressivement, troubles du rythme ou de la conduction cardiaque pouvant entrainer des morts subites.
– Atteinte rénale : syndrome néphrotique pouvant évoluer vers une insuffisance rénale terminale avec des reins de taille normale, en général sans hypertension artérielle associée.
– Atteinte digestive : perforation, hémorragie digestive, obstruction intestinale, gêne à l’alimentation et obstruction des voies aériennes du fait de la macroglossie.
– Atteinte pulmonaire : insuffisance respiratoire rapidement progressive en cas d’atteinte interstitielle touchant les bronchioles terminales et les alvéoles (lieux des échanges gazeux), plus fréquente en cas de sécrétion d’IgM.
– Complications hémorragiques : dues à l’infiltration vasculaire de l’amylose, à un déficit en facteur X (plus rarement V ou IX) ou à une augmentation de la fibrinolyse.
C’est le nombre et la sévérité des atteintes qui conditionnent le pronostic, surtout l’atteinte cardiaque : médiane de survie sans traitement de 13 mois pour l’ensemble des patients et de moins de 6 mois en cas de cardiopathie amyloïde symptomatique (responsables de la moitié des décès).
Les nouveaux traitements permettent d’obtenir une bonne réponse hématologique chez la majorité des patients avec une baisse des chaînes légères sériques responsables des dépôts. Si cette baisse est suffisante, au mieux complète, les atteintes d’organes vont s’améliorer doucement et en cas de prise en charge précoce, avant la constitution d’une atteinte cardiaque terminale, la survie est excellente.
La MAYO Clinic a établi un score pronostique permettant un classement en 3 stades en fonction de la sévérité de l’atteinte cardiaque (Figure 2).
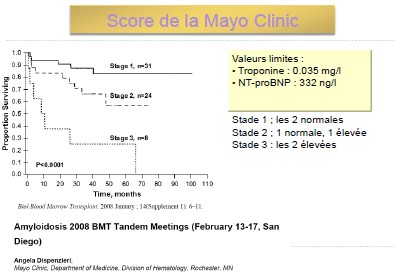
Figure 2. Score pronostique dans les amyloses AL selon la MAYO Clinic.
Traitement
Le premier but du traitement est de faire baisser le taux de la protéine monoclonale (chaînes légères le plus souvent) responsable de la formation des dépôts et d’agir sur la balance formation-élimination des dépôts par l’organisme. Si le taux des chaînes légères libres baisse suffisamment, les atteintes d’organes vont progressivement s’améliorer. La diminution des dépôts hépatiques peut être obtenue en 3-4 mois alors que la régression de l’atteinte cardiaque prendra plusieurs années. Cette baisse est obtenue par les chimiothérapies efficaces sur l’hémopathie responsable qui est le plus souvent proche d’un myélome. Les progrès dans le traitement des amyloses AL ont suivi ceux du myélome avec l’utilisation des nouvelles molécules. Le traitement initial est adapté à la sévérité des atteintes et ensuite à la réponse jugée sur la baisse du taux des chaines légères libres (Figure 3). En cas de non réponse aux associations utilisées en première ligne (Corticoïdes, Alkylants (Endoxan ou Melphalan) et Inhibiteurs du Protéasome), tous les autres médicaments efficaces dans le myélome peuvent être utilisés, en particuliers les IMIDs (Lenalidomide et Pomalidomide).
Plus récemment sont apparues des stratégies thérapeutiques très prometteuses utilisant des anticorps reconnaissant les dépôts d’amylose et capable de recruter les cellules phagocytaires pour les éliminer rapidement. Plusieurs protocoles sont en cours pour les tester.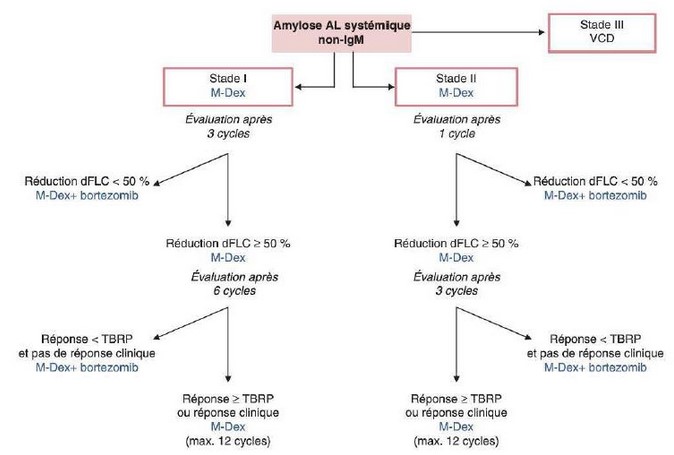
Figure 3. Traitement de l’amylose AL selon la Mayo Clinic.
Abréviations : TBRP ; Très Bonne Réponse Partielle
Les patients présentant une amylose localisée relèvent d’un traitement local, efficace dans la majorité des cas (chirurgie, laser, radiothérapie), sans indication à une chimiothérapie systémique.
Au traitement spécifique de l’amylose AL s’associe le traitement symptomatique des différentes atteintes (Tableau 4).
Organe atteint | Traitement | Prudence ou contre-indications |
Cardiaque | - Amiodarone si troubles du rythme - Diurétiques fortes doses (Furosémide) si œdèmes des membres inférieurs ou dyspnée, à moduler de façon journalière en fonction du poids +/- diurétiques thiazidiques - Pacemaker si troubles de la conduction symptomatiques ou bradycardie - Transplantation cardiaque dans les formes les plus graves avec traitement spécifique de l’amylose associé si <65ans | - IEC, inhibiteurs calciques et bétabloquants déconseillés : risque d’hypotension ou de bradycardie - Prudence : digitaliques si troubles du rythme auriculaire |
Rénale | - Diurétiques de l’anse ou épargneur de potassium si œdèmes des membres inférieurs dus au syndrome néphrotique - Dialyse si insuffisance rénale terminale - Transplantation rénale : risque de récidive de la maladie sur le greffon | - Anti-inflammatoire non stéroïdien |
Neurologique | - Hypotension orthostatique : Minodrine 2,5mg x 3/J-> 10mg x 3/J Bas de contention |
Tableau 4. Traitement symptomatique des différentes atteintes amyloïdes.
La prophylaxie anti-infectieuse comprend souvent un traitement par Bactrim Forte en cas de corticothérapie, Zélitrex en cas de traitement par inhibiteur du protéasome et Oracilline.
En cas de traitement par IMID, une prophylaxie anti-thrombotique est mise en place par Aspegic ou héparine de bas poids moléculaire en fonction des antécédents et des facteurs de risque thrombotiques du patient.
Suivi
Le suivi repose sur l’évaluation de la réponse hématologique d’une part et l’évaluation des atteintes d’organes d’autre part.
L’évaluation de la réponse hématologique est basée sur le dosage des chaînes légère libres sériques, à faire si possible dans une structure hospitalière, ces tests n’étant pas remboursés.
L’évaluation des atteintes d’organes est réalisée de la même manière que lors du bilan initial (Tableau 2 et tableau 3).
Conclusion
L’amylose AL est une pathologie pouvant se présenter de façon extrêmement diverse et de pronostic sévère en l’absence de prise en charge rapide. Le développement de nouvelles stratégies thérapeutiques a permis l’amélioration considérable du pronostic des patients atteints d’amylose AL. Un diagnostic et une prise en charge précoces sont donc primordiaux pour mettre rapidement en place une stratégie thérapeutique efficace avant la constitution d’atteintes d’organes sévères en particulier cardiaque.
Amylose Al - Pour en savoir plus
Site internet du centre de référence « Amylose AL et autres maladies par dépôt d’immunoglobulines monoclonales » : http://www.unilim.fr/cr-amylose-al/Jaccard A, Desport E, Mohty D, Bridoux F. Amylose AL. Rev Med Interne. 2015 Feb;36(2):89-97.
Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun 25;387(10038):2641-54.
- par Sammara Chaubard, Arnaud Jaccard


